闵庆祝, 王长松
(沈阳化工学院,辽宁沈阳110142)
摘 要:利用3-甲硫基丙醛与甲醛在碱性条件下所发生的坎尼扎罗反应合成新的三元醇:2,2-二羟甲基-3甲硫基丙醇,进而合成新的螺环原碳酸酯化合物:3,9-二羟甲基-3’,9’二乙硫基-1,5,7,11-四氧杂螺环[5・5]十一烷,通过红外光谱、核磁共振氢谱和元素分析表征其结构.在三氟化硼单乙胺络合物的催化作用下,该化合物能够进行阳离子开环聚合反应.用膨胀计法跟踪测试了在聚合反应过程中该化合物的体积膨胀率约为4・05 %.并且合成了该化合物与TDI反应生成的预聚物,研究了该预聚物与环氧树脂的共聚固化反应,对共聚过程中的反应历程进行初步探讨,并测试了其改性效应.
关键词:螺环原碳酸酯; 预聚物; 阳离子开环聚合反应; 环氧树脂
中图分类号:O631 文献标识码:A
文章编号:1004-4639(2008)04-0327-04
自美国化学家Bailey教授发现膨胀聚合反应以来[1],这一类型反应的研究已取得了长足的进展.由于膨胀聚合反应具有一般聚合反应所没有的特点,即在聚合过程中产生体积膨胀从而赋予了聚合物奇异的性质,引起了高分子研究人员的极大兴趣.用膨胀单体的预聚物与热固性树脂共聚,通过调节适当的比例可以获得无体积收缩的材料,从而彻底解决体积收缩这一问题.膨胀聚合反应可用于精密铸造、高强度粘合剂、密封材料、补缝材料、补牙材料等.例如,利用螺环原碳酸酯改性环氧树脂制备的碳纤维复合材料可以使材料的韧性得到明显的改善,而剪切强度和其它力学性能基本不变,甚至略有提高[2].用螺环原碳酸酯改性环氧树脂制成的一种特殊黏合剂,黏接玻璃时其光学性能基本不变[3].本文合成了新的螺环单体3,9-二羟甲基-3’,9’二乙硫基-1,5,7,11-四氧杂螺环[5・5]十一烷,研究了该螺环单体与TDI反应生成的预聚物对环氧树脂的改性效应,并测定了改性树脂材料的力学性能和热性能.
1 实验部分
1・1 试剂
2,2-二羟甲基-3甲硫基丙醇是在碱性条件下由3-甲硫基丙醛与甲醛缩合而成.二正丁基氧化锡为北京恒正产品.环氧树脂(3,4-环氧基环己烷甲酸-3’,4’-环氧基环己烷甲酯)为天津合成材料工业研究所生产.三氟化硼乙胺为上海试剂三厂产品.
1・2 仪器
红外光谱仪:IR-435,日本岛津公司产品.核磁共振仪:R-90H,日本日立公司产品.元素分析仪:菲尼根1112型,美国热电集团产品.热分析仪:美国PE公司产品.
1・3 3,9-二羟甲基-3’,9’二乙硫基-1,5,7,11-四氧杂螺环[5・5]十一烷的合成
将0・1 mol的2,2-二羟甲基-3甲硫基丙醇、0・1 mol二正丁基氧化锡一并加入250 mL三口瓶中,加入150 mL甲苯,装上分水器和回流装置,电磁搅拌,控制油浴温度130℃,反应12 h.将油浴温度降至室温,拆除分水器,装上恒压滴液漏斗和回流装置,并在回流冷凝管上安装CaCl2干燥管.在漏斗中加入CS28 mL,缓慢滴入反应瓶中,并将油浴缓慢加热到100℃,回流12 h,降至室温后,减压蒸馏出甲苯.用正己烷100 mL分5次洗涤以除去n-Bu2SnS,洗涤过程中继续搅拌,底部黏稠液体为较纯正的单体.加入甲苯50 mL,加热使其溶解,冷却至室温,干燥,即为产品,为褐色黏稠液体.
核磁共振氢谱δ:4・78(2H,―OH):3・45(4H,―CH2OH); 3・8 (8H,―O―CH2―); 3・35(4H,―S―CH2―);2・62(―S―CH3).
红外光谱:在波数1 212 cm-1出现明显的螺环基团特征吸收峰.
元素分析: C13H24S2O6,计算值(%) C:45・88;H:7・06.实测值(%)C:46・59;H:6・89.
1・4 螺环单体的均聚反应
将质量分数3 %的三氟化硼单乙胺催化剂加入到液态螺环单体中,搅匀.真空烘箱中恒温60℃脱气至无气泡.然后恒温120℃实施阳离子开环聚合反应,用膨胀计跟踪聚合过程中体积的变化,均聚物的表征结果如下:
核磁共振氢谱δ:4・78(2H,―OH);3・45(―CH2OH);3・8(8H,―O―CH2-);3・4(4H,―CH2―O―C―O―); 3・35 ( 4H,―S―CH2―);2・62(―S―CH3).
红外光谱:在波数1 212 cm-1处的螺环基团特征吸收峰消失,而在1 732 cm-1处出现了很强的羰基吸收峰,在波数1 090 cm-1处出现链状醚键的特征吸收峰.
1・5 预聚物的合成
将溶在氯仿中的TDI按不同的原料配比n(M)∶n(TDI)=2∶1,3∶2,4∶3逐滴加入到螺环单体的氯仿溶液中.滴加完毕后,在35℃反应24 h,然后将所得物质倒入表面皿中,使其自然冷却,溶剂自然挥发,放置48 h,将所得物质研磨成粉末,即得产品,为褐色固体.
1・6 预聚物和环氧树脂的共聚固化反应用DSC研究共聚固化反应的步骤如下:以不同质量配比将螺环单体和环氧树脂均匀混合,再加入三氟化硼单乙胺(树脂质量的3 %),搅匀后,放入真空烘箱中脱气2 h,以排除树脂内的气泡.称取5~10 mg树脂样品放入铝坩埚内,封好后放入样品池中,以10℃/min的升温速率,跟踪共聚固化反应的进行.
用红外光谱测定环氧基团转化率的方法如下:将配好的树脂样品均匀地涂在溴化钾盐片间,然后放在140℃烘箱中固化4 h,在岛津IR-435上测试固化前后的红外光谱.根据环氧基团在913 cm-1处的特征吸收峰高(A913)的高低,以1 610 cm-1处苯环骨架振动吸收峰高(A1 610,在聚合反应过程中保持不变)作为标准,按下式计算环氧基团的转化率(α)[4]:

1・7 黏接强度的测定
将试样(45#钢)表面打磨光滑,用丙酮洗涤,然后把配比不同的改性环氧树脂均匀地涂在表面后,将两个试样胶接在一起,将其牢牢固定好后,放入恒温在120℃的烘箱中固3 h.慢慢冷却到室温后,在万能材料试验机上测试其抗拉强度和剪切强度,夹头间距为50±2 mm,拉伸速率为6 mm/min,取5个试样平行测试结果的平均值.
2 结果与讨论
2・1 体积膨胀率的测定
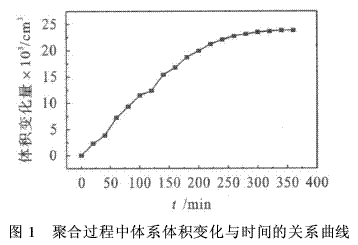
实验合成了新的螺环单体:3,9-二羟甲基-3’,9’二乙硫基-1,5,7,11-四氧杂螺环[5・5]十一烷,用膨胀计跟踪测量聚合过程中体系体积的变化,见图1.由测量结果可求得聚合前后的体积变化率为:
α=ΔV/ V=πr2Δh/(W/d)×100 %=24・52×10-3×1・192 4/0・721 19×100 %=4・05 %
式中r为毛细管半径;d为单体密度.
2・2 预聚物的结构表征
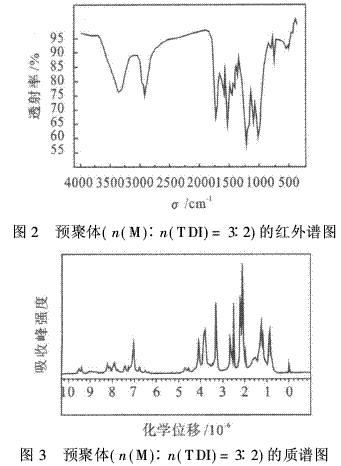
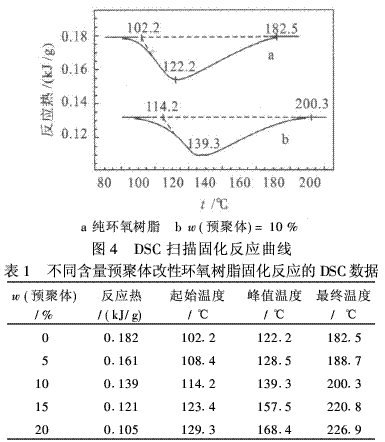
红外光谱和核磁共振氢谱分别见图2和图3.由图2可知,1 217 cm-1处螺环基团的特征峰仍然存在,1 731 cm-1处出现氨基甲酸酯基中的羰基吸收峰;3 346 cm-1处出现了N―H的吸收峰;1 534 cm-1处出现了苯环的吸收峰.由图3可看出,在3・1×10-6处是羟质子峰,3・2×10-6处是胺基质子峰,3・4×10-6处是与羟基相连的亚甲基吸收峰,3・8×10-6处是螺环上的亚甲基质子峰,4・2×10-6处是与胺基甲酸酯键相连的亚甲基峰,7・0~7・6×10-6处是苯环的质子峰.并且峰面积的积分与各种质子数的比例相对应.综上所述,表征的结果与预聚物的结构是一致的.
2・3 预聚物与环氧树脂的共聚固化反应
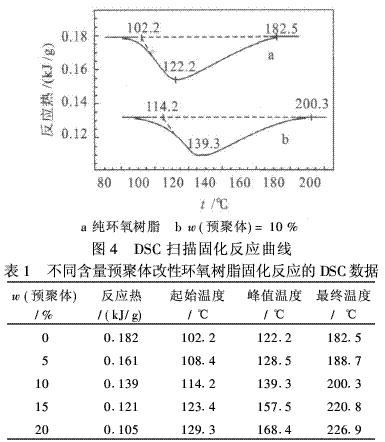
使用一种潜伏性的催化剂:三氟化硼单乙胺,其分解温度为90℃,分解时放出催化活性种.这样做的好处,一是容易进行前期预处理工作;二是在该温度下,环氧基团和螺环基团能够同步开环,进行共聚固化反应,因为环氧基团的开环聚合活性比螺环基团要高[5].跟踪共聚固化反应进行的DSC曲线也证实了这一点,即加有螺环单体的改性环氧树脂的反应热曲线向高温方向移动,如图4所示.由图4可见,加有螺环单体的改性环氧树脂的固化反应放热曲线与纯环氧树脂的相似,只出现1个放热峰,但随着加入量的增加,放热峰面积减小,即放热量减小,峰的位置也向较高温方向移动(见表1),这说明预聚物的反应热与环氧树脂相比要小.
由于二异氰酸酯(TDI)中的苯甲烷基与环氧树脂基体的相容性较好,使得环氧基团和螺环基团之间能够较好地共聚.并且,在相同的固化反应条件下,环氧基团的聚合速率大于螺环基团,当反应开始时,以环氧基团之间的反应为主;当交联结构形成后,环氧基团的活动性受到了限制,而预聚物此时仍有相当大的活动性,它可以与环氧基团碰撞而进行共聚合反应,生成支链或交联结构;或螺环基团自聚,形成互穿网络结构.由此可见,预聚物参与了环氧树脂的固化反应,从而能够影响环氧树脂的结构和性能[6].
2・4 黏接强度分析
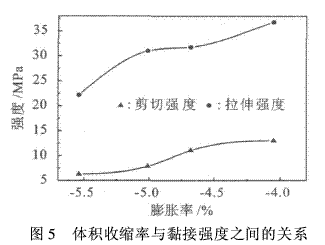
由于热固性高分子材料在固化时体积收缩,限制了它在某些特殊场合下的应用.用膨胀性螺环单体对其改性以制备非收缩或稍微膨胀的高分子材料是一个重要的应用方向,所以考察了螺环原酸酯的加入量对黏接抗拉强度和黏接剪切强度的影响,结果见表2.
结果表明,随着加入量的增加,其抗拉强度和剪切强度均呈上升趋势,特别是剪切强度上升更为明显.其原因可能是,螺环原酸酯的加入减少了聚合过程中的体积收缩率,特别是环氧树脂交联结构基本形成以后,体积收缩率的降低可以大大降低胶接件之间的收缩应力,所以随着加入量的增加,体积收缩率减少,导致环氧树脂的黏接强度增加,试验也证实了这一点(见图5);但当螺环原酸酯加入量较大时(例如20 %),黏接强度有所降低,一方面是由于此时树脂体系的黏度较大,对试件表面浸润性不良而引起的;另一方面是由于预聚体含量过大,在改性环氧树脂内部的均匀分散很困难,容易集聚,从而造成缺陷,使得改性环氧树脂的强度下降.
参考文献:
[1]Bailey W J,Sun R L.Polymerization of a Spiro OrthoEster[J].Amer
Chem Soc Div Polym Chem Pre,1972,13(1):281-286.
[2]Piggott M R,Bailey W J.Dental Resins with Re-duced Shrinkage
During Hardening[J].Sampe Quar-terly,1984,7:25.
[3]潘才元,周志强,何平笙.光学玻璃的胶接试验[J].粘接,1986,7(5):77.
[4]Feazel C E,Verchot E A.Simple Technique for Usewith Infrared
Spectroscopy to Study the Curing of E-poxy Resins[J].J Polym
Sci,1957,25:351.
[5]白如科,王长松.含螺环原碳酸酯齐聚物的合成、表征及与环氧树脂的固化反应[J].功能高分子学报,1995,8(3):321-327.
[6]王长松,潘才元.螺环原碳酸酯的预聚物对环氧树脂的改性研究[J].沈阳化工学院学报,1996,10(2):87-91.
