|
张 斌, 孙明明, 张绪刚, 王 超, 李奇力, 关长参
(黑龙江省石油化学研究院,黑龙江哈尔滨 150040)
摘要:本文用二元醇M2与MDI反应(-NCO∶-OH=2∶1),合成端-NCO基预聚体;然后用预聚体与环氧树脂E-51反应,合成聚氨酯改性环氧树脂。由FTIR研究表明,改性后环氧基团未发生变化;由SEM研究表明,随着加入量的增大,聚氨酯橡胶逐渐聚集,改性体系的微观形态发生了三种不同的变化。受微观形态影响,改性体系的力学性能和粘接性能也发生了变化。本文对不同固化体系的粘接性能也进行了探讨,结果表明改性体系具有优异的粘接性能。
关键词:环氧树脂;聚氨酯;改性;微观形态,力学性能
前 言
环氧树脂是一种热固性树脂,因具有优异的粘接性、机械强度、电绝缘性及良好的工艺性等特性,而广泛应用于胶粘剂、涂料、复合材料基体等方面。由于纯环氧树脂具有较高的交联结构,因而存在质脆、耐疲劳性、耐热性、抗冲击韧性差等缺点,难以满足工程技术的要求,使其应用受到一定的限制,因而对环氧树脂的改性工作一直是中外研究的热门课题,其中环氧树脂的增韧是改性工作中的一个重要领域[1-5]。
环氧树脂的增韧方法很多,虽然研究探讨了几十年,有些增韧方法已经很成熟,但近来仍不断有新的增韧方法、新的增韧剂出现。目前国内外的研究主要集中在两方面,一方面是随着技术的不断发展,如何获得具有更高性能的高分子材料,以满足许多特殊场合的要求,并能使其得到更广泛的应用;另一方面是随着市场的不断发展,如何能够获得具有更低成本的高分子材料,以适应市场的需求。CTBN是非常成熟、增韧环氧树脂性能非常优异的品种,但是由于其昂贵的价格和较高的粘度使其只能在少数场合使用,限制了其应用。如何解决性能与成本等之间的矛盾,始终是国内外研究的重点。
聚氨酯是一类性能优良的高分子材料,尤其具有高弹性、高粘接力的优点,是近年来发展迅速的高分子材料之一。它可与环氧树脂以多种方式结合,并展现出各自的优点。聚氨酯产品价格低廉,如果能够改性环氧树脂,获得优异的性能,并且能够有良好的工艺性能,那么市场应用的前景非常广阔[6-14]。
1 实验部分
1.1 实验原料
组合聚醚M2(分子量2000,官能度2),自制;环氧树脂E-51,工业品,无锡树脂厂;4,4’-二氨基二苯甲烷(MDA),分析纯,上海试剂一厂;室温固化耐温固化剂A,本实验室合成;二苯基甲烷-4,4’-二异氰酸酯(MDI),工业品,烟台万华。
1.2 实验方法
剪切试片:化学氧化处理
剥离试片:磷酸阳极化处理
拉伸、冲击试片:按GB2567-81自制
剪切强度:GB 7124-86
剥离强度:GB 7122-86
弯曲冲击强度(胶粘剂):胶粘剂冲击强度通用方法
拉伸强度、拉伸杨氏模量、断裂伸长率:GB 2568-81
弯曲强度、弯曲杨氏模量:G B2570-81
冲击强度(树脂浇铸体):GB 2571-81
红外谱图由Nicolet 50X测定
扫描电镜由JSM-840测定
1.3 聚氨酯改性环氧树脂的合成
预先将M2加热到100-140℃,抽真空,保持真空度为10~100mmHg,减压脱水2-4小时。将MDI加入三口瓶中,通N2气保护,搅拌升温至80℃。按比例加入预先加热好的多元醇(-NCO∶-OH为2∶1,-NCO基过量5%),反应2小时后,按比例加入预先加热好的E-51继续反应。反应2小时后,加入1%的二丁基二月桂酸锡,继续反应0.5小时后停止反应,产物倒出。
2 结果与讨论
2.1 聚氨酯改性环氧树脂的表征
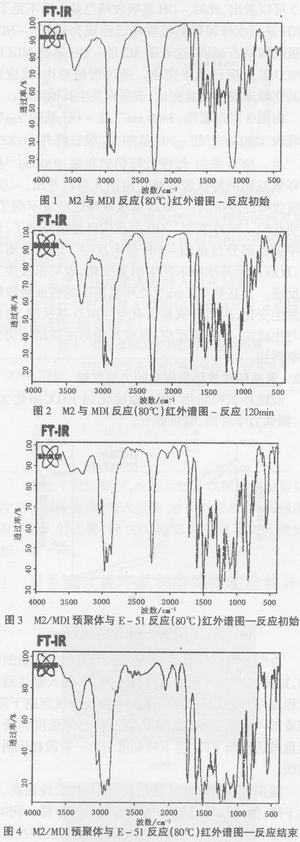
由图1可以看出,在3480cm-1处为-OH基的γ(OH)吸收峰;在2270cm-1处-NCO基的γas吸收峰非常明显;1730cm-1出现了酯基的γc=0吸收峰,3280cm-1处也有微弱的-NH基的γNH吸收峰出现。这表明-NCO基的活性非常高,此时已有少量反应。由图2可以看出,此时-OH基吸收峰己基本看不见了,-NH基吸收峰和羰基吸收峰已经成为强峰,-NCO基吸收峰略有减弱,这表明M2的-OH基与MDI的-NCO基反应已基本完全。通过谱图对比,反应前后其它峰未发生明显变化,表明未发生其他反应。
由图3可以看出,3480cm-1处-OH基的γOH吸收峰和3280cm-1处-NH基的γNH吸收峰并存;2270cm-1处-NCO基的γas吸收峰仍然很强;920cm-1处是环氧基团的特征吸收峰。由图4可以看出,-OH基吸收峰已经很弱,-NCO基吸收峰已经看不到了,说明此时-NCO基与-OH的反应已经很完全了,体系中仍有部分过量的-OH基未反应。通过谱图对比,反应前后其他峰未发生明显变化,表明未发生其他反应。尤其是920cm-1处环氧基团的特征吸收峰未发生变化,表明环氧基没有与-NCO基反应,也没有与生成的-NH基反应,反应产物的主体结构仍为环氧树脂。
2.2 聚氨酯改性环氧树脂的力学性能
改性环氧树脂用MDA做固化剂100℃固化3 h后,测试力学性能,结果如下:
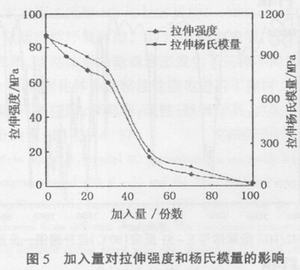
由图5可以看出,随着聚氨酯预聚体加入量的增加,拉伸强度和杨氏模量一直下降;在加入量达到一定程度(30份)以后,拉伸强度和杨氏模量急剧下降;随着加入量继续增加(50份以后),拉伸强度和杨氏模量虽仍逐渐下降,但下降幅度与上一阶段相比明显减缓。
随聚合物中柔性链段增加,聚合物柔性提高、刚性下降,因此拉伸强度和杨氏模量一直下降。同时,体系固化后产生了微观相分离,聚氨酯橡胶微粒以分散相分散在环氧树脂的连续相中。在不同阶段橡胶相产生的作用也不相同,加入量较小时,橡胶相起到受冲击时引发银纹并阻止银纹进一步发展,吸收能量的应力集中物的作用,总体上仍表现出环氧树脂连续相的性质,因而拉伸强度和杨氏模量虽因柔性链节的引入而下降,但下降幅度相对较小;在加入量达到一定程度(30份)以后,由于聚氨酯橡胶粒子聚集,粒径逐渐增大,相分离程度加深,橡胶相类似于体系的缺陷,体系主导作用表现为环氧树脂连续相被破坏,因而拉伸强度和杨氏模量大幅下降;随着加入量继续增加,聚氨酯逐渐聚集形成连续相,体系逐渐表现出了聚氨酯的性能,拉伸强度和杨氏模量虽下降,但下降幅度与上一阶段比有所缓和。
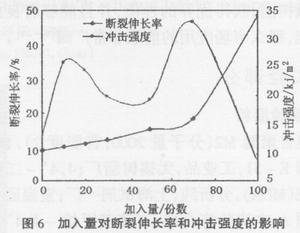
由图6可以看出,随着聚氨酯预聚体加入量的增加,断裂伸长率不断增加。在加入量达到一定程度(70份)以后,断裂伸长率迅速增加。随着预聚体加入量的增加,冲击强度不断增加,在加人量为10份时出现第一个峰值,然后随着预聚体加人量的增加冲击强度开始下降;在加入量达到50份后,冲击强度又开始增加,在加入量为70份时出现第二个峰值,然后冲击强度又开始下降。
随聚合物中柔性链段增加,聚合物柔性提高、刚性下降,因此断裂伸长率一直增加;在体系的性能向聚氨酯的性能转变后,断裂伸长率迅速增加。
同样由于体系柔性的增加及橡胶相在不同阶段所起作用不同,冲击强度的变化出现了二个峰值。在橡胶相以增韧作用为主时,冲击强度增加;在橡胶相
以破坏主固化体系作用为主时,冲击强度下降;聚氨酯形成连续相后,冲击强度又增加;加入量太高时,由于体系变软了,冲击强度又下降,在100份时,强度比纯环氧树脂还要低。此外,70份和100份时,冲击试片呈坚韧的橡胶状,受冲击时弯曲,打不断。
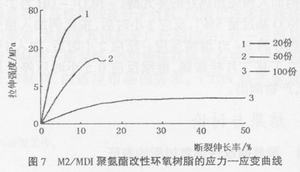
由图7可以看出,聚氨酯橡胶相含量较小时(20份),破坏形式表现为脆性断裂;在聚氨酯橡胶相含量达到一定程度(50份),应力-应变曲线上出现屈服点,破坏形式表现为韧性断裂;在聚氨酯橡胶相含量较高时(100份),破坏形式表现为延性断裂。实际上,虽然橡胶加入量为50份时,破坏形式为韧性断裂,但此时拉伸强度已经很低了。
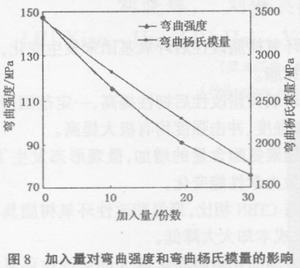
由图8可以看出,随着聚氨酯预聚体加入量的增加,弯曲强度和弯曲模量一直下降,这也是体系刚性下降的结果。此外,聚氨酯预聚体添加至50份后,固化后的树脂太软,弯曲强度、弯曲杨氏模量都测不出来。
2.3 聚氨酯改性环氧树脂的粘接性能
2.3.1 MDA固化体系
改性环氧树脂用MDA做固化剂100℃固化3 h后,测试粘接性能,结果见图9。
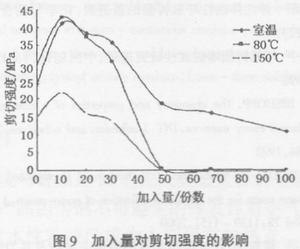
由图9可以看出,改性后的环氧树脂脆性得到了改善,在加入量较小时剪切强度有了明显的提高,加入量为10份时最高,随后开始下降,尤其是80℃和150℃剪切强度在加入量超过50份时已经非常低了。
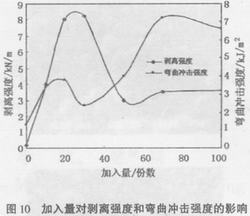
由图10可以看出,改性后的环氧树脂剥离强度和冲击强度有了明显的提高。加入量为20份时冲击强度最大,然后开始下降;超过30份以后冲击强度又开始提高,在70份时达到最大,然后又开始下降。加入量为30份时剥离强度最大,然后急剧下降,在50份时达到最低,然后逐渐提高。
这也是由于随加入量的增加,改性后的环氧树脂由环氧树脂连续相、聚氨酯分散相的相分离状态向双连续相过渡产生的结果。
2.3.2 室温固化耐温固化剂A固化体系
改性环氧树脂用A做固化剂室温固化7d后,测试粘接性能,结果如下:
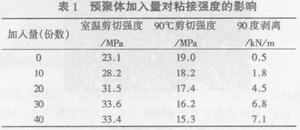
由表1可以看出,改性后的环氧树脂室温剪切强度有了很大提高,加人量为30份时最高;剥离强度也有了极大的改善;但90℃剪切强度随着加入量的增
加一直下降。
3 聚氨酯改性环氧树脂的微观结构分析
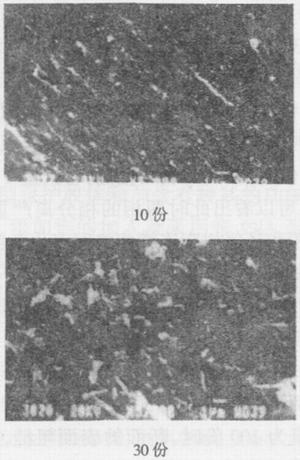

图11分别为M2/MDI体系,预聚体加入量分别为10、30、50、70和100份时,用MDA固化后弯曲断裂表面的SEM照片(5000倍)。从照片中可以看出,随着预聚体加入量的增加,改性体系断裂面的形态发生了明显的变化。当预聚体加入量为10份时,断面平整、光滑,断裂后很难看出橡胶颗粒脱出后的孔穴,可
以看出此时的断裂形式仍为脆性断裂。当预聚体的加入量为30份时,断面的表面粗糙,两断面之间的相互穿插十分明显,而且可以观察到部分直径为0.5μm左右的橡胶颗粒脱出后留下的“孔穴”状情形,可以看出此时的断裂形式为韧性断裂。当预聚体的加入量为50份时,断面的表面粗糙,有大量1-3μm球状物析出,可以看出此时两相的相分离严重;由于聚氨酯橡胶相和环氧树脂连续相之间有化学键连接,析出的可能不单单是聚氨酯橡胶相,否则也不能有这么多,析出的应该是聚氨酯橡胶相包容了一部分环氧树脂相。当预聚体的加入量为70份时,断面的表面粗糙,析出的球状物粒径减小,约为0.5-1.5μm,可以看出此时两相的相分离较50份时有所改善。当预聚体的加入量为100份时,断面的表面粗糙,析出的球状物粒径进一步减小,约为0.2-1μm,此时两相的相分离较70份时进一步改善。
随着预聚体加入量的增加,橡胶粒子不断聚集,改性体系逐渐由均相向两相过渡,相分离程度不断增加,在达到一定程度时,两相分相严重。预聚体加入量继续增加,聚氨酯橡胶相逐渐向连续相转变,两相的互溶性又逐渐得到改善。
4 结 论
1、环氧树脂改性后环氧基团未发生变化,主体仍是环氧树脂。
2、环氧树脂改性后韧性提高,一定范围内剪切强度、剥离强度、冲击强度均有极大提高。
3、随聚氨酯含量的增加,微观形态发生了变化,进而导致力学性能变化。
4、与CTBN相比,聚氨酯改性环氧树脂具有相近的性能,成本却大大降低。
5、室温固化耐温固化剂A固化改性环氧树脂具有优异的粘接性能,良好的工艺性能,可在多种场合得到应用。
参考文献:
[1] BYOUNG UN KANG,Effect of Molecular Weight between Grosslinks on the
Fracture Behavior of Rubber-Toughened Epoxy Adhesives,J Appl Polym
Sci,Vol 79,38-48,2001
[2] A J KINLOCH.Deformation and fracture behaviour of a rubber-toughened
epoxy:1.Microstructure and fracture studies,Polymer,vol
24,1341-1354,1983
[3] 欧召阳.弹性体改性环氧树脂的新进展,化学与粘合,2001(3);12-123.
[4] 刘志中,环氧树脂增韧改性研究进展,中国塑料,Vol 12(6),12-17,1998
[5] J A BISHOPP.The chemistry and properties of a new genetation of
toughened epoxy matrices,INT.J adhesion and adhesives,Vol
12,178-184,1992
[6] J KARGER-KOCSIS.Use of Hydrothermal Decomposed polyester-Urethane
waste for the impact modification of epoxy resins,J Appl Polym Sci,Vol
78,1139-1151,2000
[7] 茅素芬,聚氨酯/环氧树脂共混物的形态结构及其力学性能,高分子材料科学与工程,Vol 12(1),85-90,1996
[8] H.HARANI,Toughening of Epoxy Resin Using Hydroxyl-terminated
polyesters J Appl Polym Sci,Vol 71,29-38,1999
[9] TONGHUI CHEN,Study on relationship between Mechanical Properties and
Particle Size Distribution for Polyhexamethylent Carbonate Diol
Toughened Epoxy Resin,J Appl,Polym Sci,Vol 67,569-575,1998
[10] HUEI-HSIUNG WANG,Toughening of epoxy Resin by Functional-
Terminated Polyurethanes and/or Semicrystalline Polymer Powders,J
Appl Polym Sci Vol 82,2903-2912,2001
[11] P.M.STEFANI,Epoxy-Urethane Copoymer:Relation Between Morphology and
droperties,J Appl Polym Sci,Vol 82,2544-2522,
2001
[12] TONGHUI CHEN, Study on epoxy resins Modified by
polyearbonate polyurethanes, J Appl Polym Sci, Vol 69,887 - 893,1998
[13] H. HANRANI, Toughening of Epoxy Resin Using Synthesized
Polyurethane prepolymer Based on Hydroxyl - Terminated
Polyesters,J Appl Polym Sci,Vol 70,2603-2618,1998
[14] PABLO M STEFANI,Polyurethane-Ductilized Epoxy Resins,J Appl Polym
Sci,Vol 68,1781-1789,1998
|
